

Luis Gosálbez & Andrea Almagro
INTRODUCTION
Every corner of the human body is inhabited by microbes, collectively known as the microbiome. Thousands of publications over the last decade have described how these microorganisms influence human physiology, health and disease, and hundreds of companies have been founded to develop microbiome-based pharmaceuticals.
The capacity of medicines, especially antibiotics, to alter the composition and activity of the microbiome has been known for long1,2. However, significantly less attention has been put on how intra- and inter-individual microbiome composition and activity affect drug action, temperament, effectiveness, and toxicity. This discipline studying the relationship between microbiomes, pharmacokinetics (PK) and pharmacodynamics (PD) is known as pharmacomicrobiomics3,4.
PURPORTED MECHANISMS IN PHARMACOMICROBIOMICS
Research shows that microbiomes in different locations (the intestine and elsewhere) interact with drugs in multiple ways (from very direct to more indirect), resulting in alterations of a pharmaceutical’s bioavailability, efficacy and safety profiles. Broadly speaking, these microbiome-drug interactions may be clustered into four categories:
- Biotransformation, which involves the direct chemical alteration of a medicine by microbial enzymes.
- Metabolic interference, where chemicals resulting from the activity of the microbiome indirectly interfere with host drug metabolism.
- Bioaccumulation, or the retention or uptake of drugs by members of the microbiome.
- Other mechanisms in which microbiome composition or activity has been shown to result in different treatment outcomes. These relationships are largely unknown, although complex interactions with the immune system, modulation of the host’s gene expression and neoantigen-like microbial molecular mimicry have been suggested as potential mechanisms5–7.
KNOWN MICROBIOME-DRUG INTERACTIONS
- Biotransformation
Biotransformation refers to a direct alteration of the chemical structure of a drug by the action of certain enzymes from members of a microbiome, which can lead to different consequences of the fate and activity of a medicine.
Microbial activation of pro-drugs into active forms was one of the first known examples of microbiome-drug interactions. As early as in the 1930s, it was described that the pre-antibiotic-era antimicrobial drug prontosil (which active antibacterial metabolite is sulphanilamide) is reduced not only by hepatic azoreductases, but also by microbiota azoreductase enzymes, leading to an increase on its efficacy8,9.
A similar case is found in aminosalicylates (also known as 5-ASA drugs) such as sulfasalazine, widely employed in Inflammatory Bowel Disease (IBD). These drugs chemically combine their active (5-ASA) with other moieties, from which they need to be separated to elicit their anti-inflammatory effect. Microbial azoreductases are also the enzymes responsible for this transformation, which means that they are in fact activated by microorganisms rather than by human metabolism.
Microbial biotransformation does not always lead to a beneficial effect, though. For example, recent research shows that, once released by microbial activity, 5-ASA can be inactivated by enzymes such as thiolases and acyl-CoA N-acyltransferases from the microbiome10.
Another well-known example of inactivation of a drug by the gut microbiome is digoxin. This medicine, used in the treatment of chronic heart failure, is designed to reach cardiovascular cells to increase their contractility11. However, members of the gut microbiome can reduce its lactone ring, converting it into its inactive metabolite, dihydrodigoxin, dramatically reducing its bioavailability and activity12.
A very paradigmatic example of how microbes can alter drug metabolism is the fate of levodopa, a drug for Parkinson’s Disease, in the gut. In order to elicit its therapeutic action, this molecule needs to be absorbed in the intestine and travel to the brain, where it is decarboxylated by human enzymes to release dopamine. It is known for long, though, that human digestive decarboxylases can break down levodopa in the gut, preventing it from reaching its target site. This is the reason why levodopa is routinely combined with carbidopa, which inhibits these human intestinal decarboxylases, in turn enabling for lower dosing of levodopa and reducing its side effects13. However, multiple members of the gut microbiome do also express decarboxylases which are not inhibited by carbidopa, and which are able to catalyze levodopa breakdown, thus reducing its bioavailability14–19. In recent years, different groups are looking for adjuvant therapies to inhibit microbial decarboxylases in order to minimize this microbial inactivation of levodopa20.
Multiple gut microbiome biotransformations have already been described, and oncology therapy has captured significant attention. For instance, in chemotherapeutic agents like 5-Fluorouracil (5-FU) and irinotecan. In the former case, microbial dihydropyrimidine dehydrogenases yield a less active form of the drug21. In the latter, microbial β-glucuronidases release a more toxic chemical species22. As in the case of levodopa, strategies are being explored to inhibit these enzymes to mitigate their clinically-deleterious impact on drug metabolism22–24.
Most pharmacomicrobiomics studies have focused on the gut. However, microbiomes from other locations have also been suggested to interfere with pharmaceuticals. Here, again, the most prominent examples are related to cancer therapy.
Gemcitabine is an antineoplastic agent used to treat different types of cancer. For this drug to elicit its therapeutics effect, it needs to be taken up by the malignant cells and converted by tumor (human) deaminases. However, if the tumor is infiltrated by intratumoral bacteria, which according to multiple authors is not uncommon in different kinds of cancer25–28, these microbes can inactivate gemcitabine with their own deaminases before it reaches its target tumor cells. Adjuvant therapy directed towards inhibiting these bacterial enzymes29 or the transporter proteins these microbes use to take gemcitabine up30 has shown some preliminary promise in lowering tumor resistance to gemcitabine.
B. Metabolic interference
Microbiome-drug interactions can be overwhelmingly more complex than mere enzymatic modification of medicines. For example, for unclear reasons, higher levels of gut-microbiome-derived butyrate enhance the therapeutic effect of the chemotherapeutic agent oxaliplatin31 and increase the sensitivity of colon cancer cells to irinotecan32. Also, urolithin A, a metabolite resulting from the transformation of tannin compounds from foods by the gut bacteria, has been shown to improve the efficacy of 5-FU33. On the other hand, urolithin B has been linked to increased toxicity of the cisplatin and paclitaxel34. However, microbiome-drug metabolic interferences can be extremely intricated. Most are completely unknown, although some mechanistic insights exist.
The toxicity of the widespread analgesic and antipyretic paracetamol (also known as acetaminophen) can be precited by examining the levels of a urinary metabolite before drug administration35. This metabolite is p-cresol, a phenol-derivative that is generated from the metabolism of aromatic amino acids by certain species of gut-dwelling bacteria, and that, importantly, humans cannot produce by themselves. Once microbes produce p-cresol in the intestine, it is absorbed and transported by the bloodstream to the liver, where it needs to be detoxified through an o-sulfonation reaction before being excreted in urine. Nevertheless, paracetamol and p-cresol are structurally similar, so both compete for detoxification by the same reaction in the liver. If p-cresol-generating microbial activity is high, circulating p-cresol levels saturate hepatic sulfonation capacity, making the liver more prone to paracetamol toxicity35.

C. Bioaccumulation
A more recently described mechanism by which the microbiome can affect drug fate is known as bioaccumulation.
It has been observed that certain microbes internalize drugs, storing them intracellularly for some time, before chemically modifying them, or even without modifying them at all. This phenomenon has been shown to reduce or delay the bioavailability and therapeutic effect of certain drugs such as cholesterol-reducing simvastatin and the widely-used antidepressant duloxetine36,37. An intriguing insight obtained with this antidepressant is that its bioaccumulation by bacteria does not alter their growth, although it does alter their behavior (measured as metabolite secretion profile).
Early experimental evidence suggests that multiple medicines from different classes may be subject to bioaccumulation.
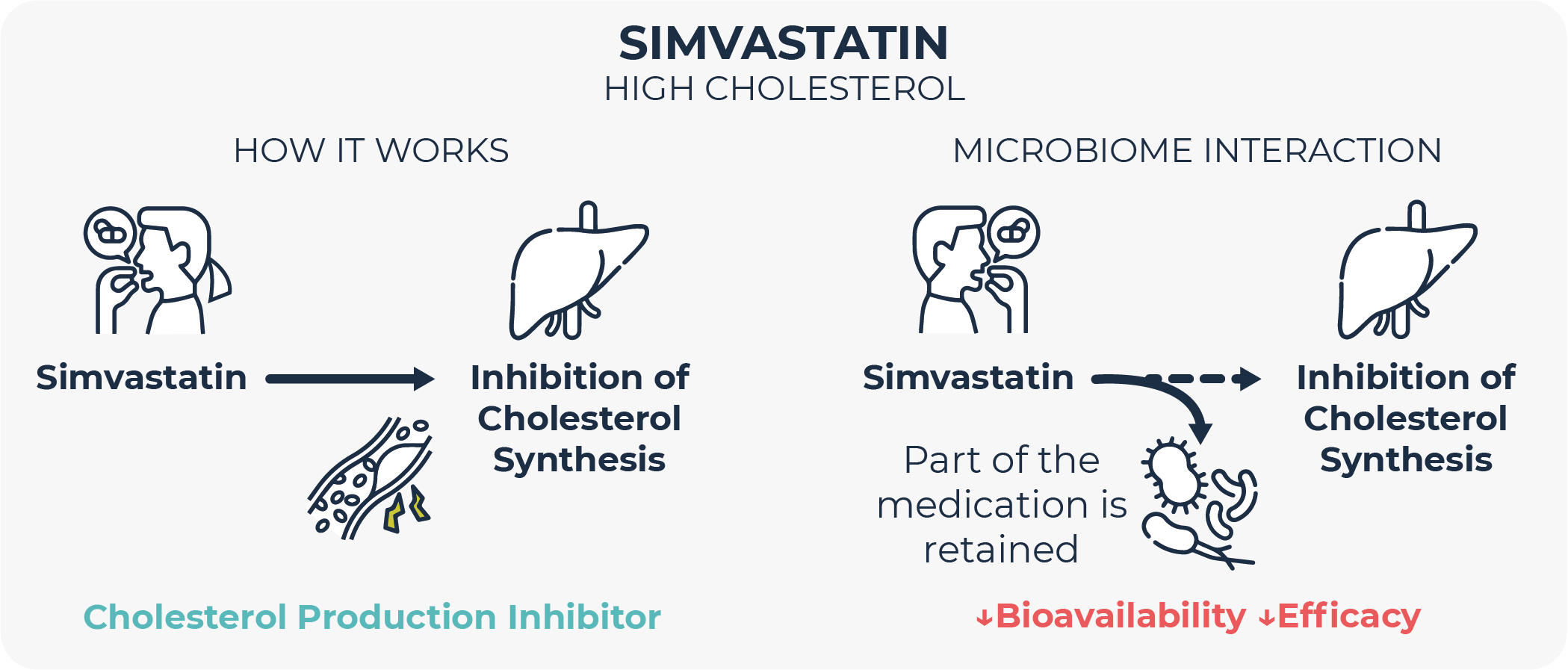
D. Other mechanisms
The gut microbiome has for long been suggested to play a role in carcinogenesis and changes in the microbiome have been observed in parallel with cancer progression38,39. However, clinical and preclinical studies over the last years have shown that microbiome composition may profoundly affect the success of immune-oncology therapy.
One of the first reports on the microbiome-cancer therapy relationship was published in 2015. In this study, researchers observed that germ-free mice or animals in which the microbiome had been depleted with oral antibiotics, did not respond to treatment with the anti-CTLA-4 monoclonal antibody ipilimumab. Also, that the composition of and the changes in the gut microbiome were correlated with ipilimumab treatment success in human melanoma patients40. Later studies confirmed that microbiome composition does not only influence treatment efficacy but also its side effects41,42. Similar observations have been reported in different types of malignancies beyond melanoma, such as colorectal and liver cancers, and with other biologics like anti-PD-L1 therapy43–45. All this has led to a great deal of interest from many pharmaceutical companies in identifying microbiome-based biomarkers for the development of companion diagnostics46 as well as of strategies to engineer the microbiome as an adjuvant treatment with biologics and cell therapies.
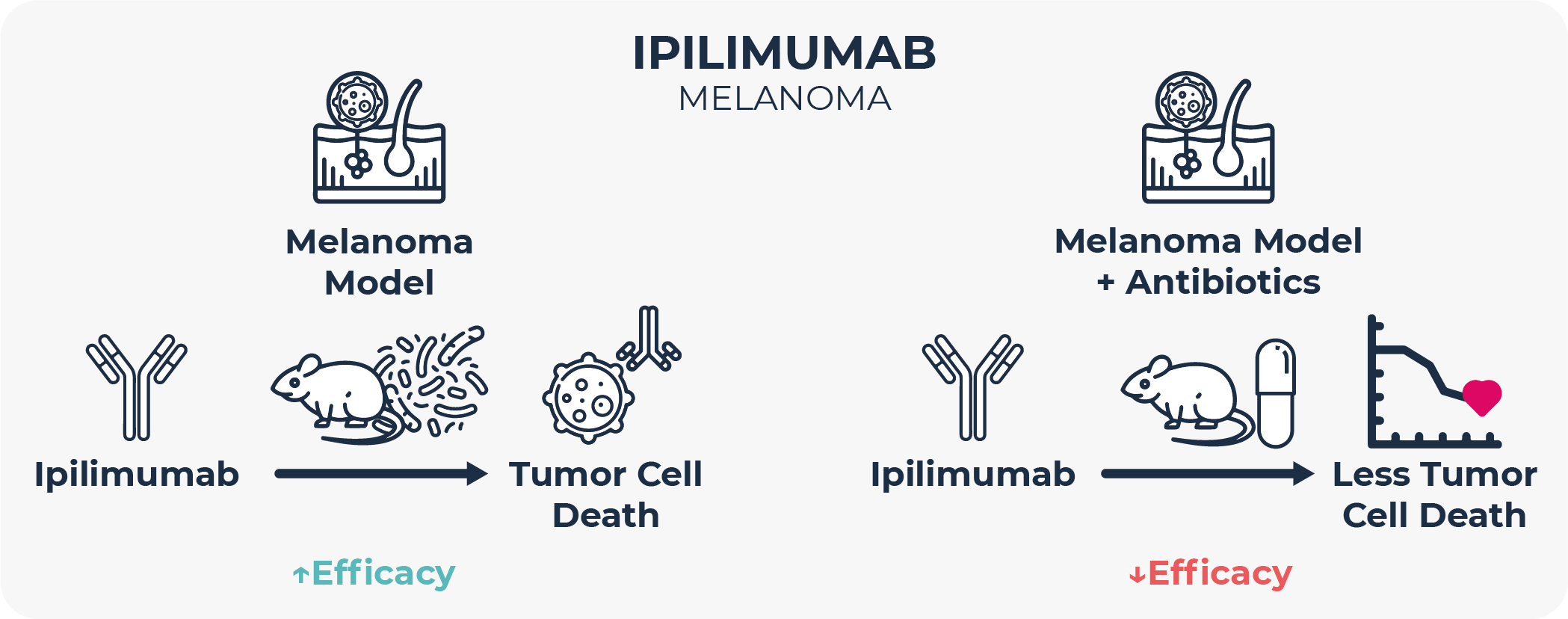
Mechanisms for these phenomena are unknown, and there is no consensus about which microbiome composition is the right one for optimal immunotherapy outcomes, although a more diverse intestinal microbial community is generally associated with better clinical response 40,43–45,47. In the absence of mechanisms, some authors argue that microbiome makeup and oncology treatment success is correlational rather than causational.
In any case, many of these studies highlight that antibiotic exposure prior to immunotherapy is linked to poorer clinical outcomes in terms of response rate, progression-free survival, overall survival, and side effects, and that therefore antibiotic use in cancer patients should be avoided as much as possible47,48. In fact, this observation has also recently extended to Anti-CD19 chimeric antigen receptor (CAR) T cell therapy, at least in B-cell lymphoma and leukemia49. Most studies hypothesize that these differences in clinical outcomes are microbiome-mediated, that is, that the changes in the gut microbiome induced by antibiotic treatment are responsible for the distinct treatment outcomes.
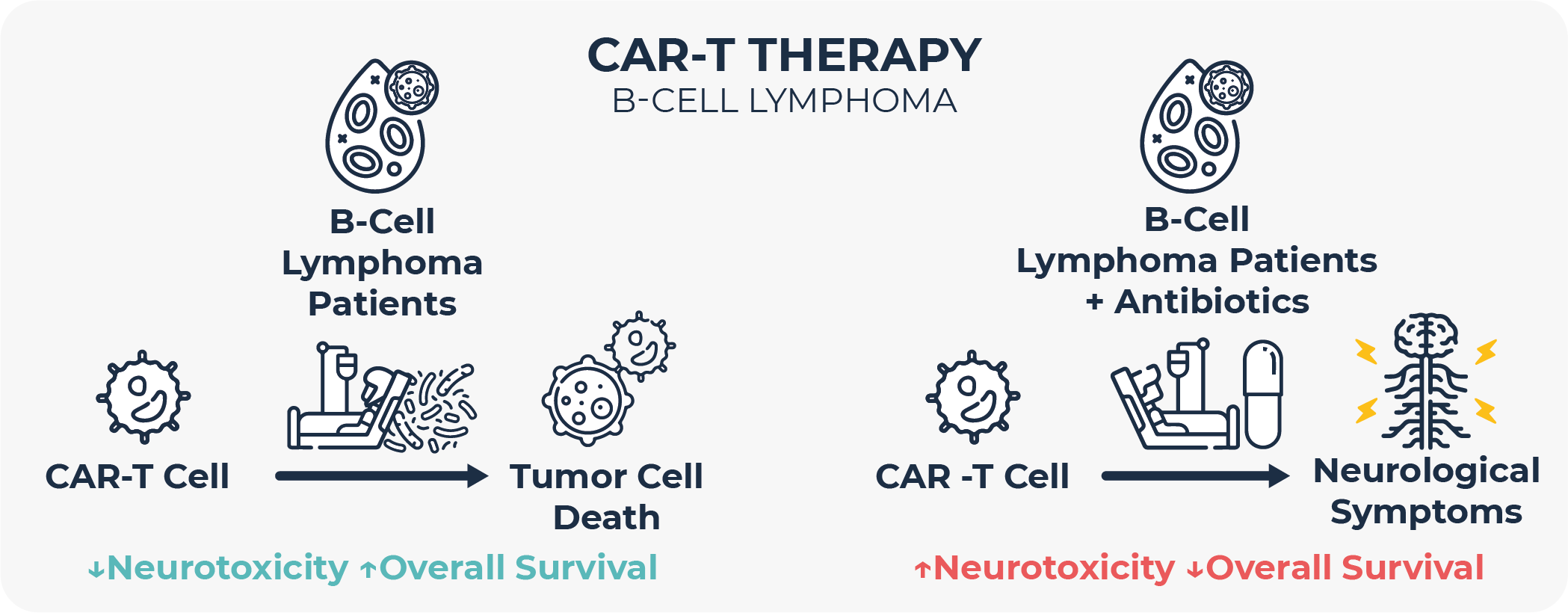
IMPLICATIONS AND FUTURE OUTLOOK
Little is still known about the extent and the mechanisms of the complex relationship between the microbiome and drugs. However, many hints point in the direction that they do not only exist, but that they are an important factor in how drugs act and in the way the human body responds to them.
It should not be forgotten that the cost of taking a medicine to market has almost doubled every nine years over the last 70, with estimates ranging between USD 2.6 and up to 6 Bn per new drug50–52, which is known as Eroom’s Law (Moore’s Law spelled backwards)53. Today, only around 8% of drug candidates entering Phase 1 trials are ultimately approved for commercialization54, with the main reasons behind failures being lack of efficacy (30%), unacceptable clinical safety and toxicity (30%), and poor pharmacokinetics (10%)55.
As described above, a better understanding of pharmacomicrobiomics may help to partially solve all these challenges and change the way drugs are thought of and designed. Knowing how bodily microbes, and how the microbiome changes associated with disease progression affect drug metabolism and action will also be crucial to the development of personalized medicine.
In the future, the microbiome will become just one more integral part in drug development, in PK/PD studies and in patient stratification, and microbiome-engineering strategies will be developed to optimize clinical outcomes of many different therapies.
References
- Vich Vila, A. et al. Impact of commonly used drugs on the composition and metabolic function of the gut microbiota. Nat Commun 11, 362 (2020).
- Willyard, C. When drugs unintentionally affect gut bugs. Nat Rev Drug Discov 17, 383–384 (2018).
- Aziz, R. K. Interview with Prof. Ramy K. Aziz, Cairo University. The Dawn of Pharmacomicrobiomics. OMICS 22, 295–297 (2018).
- Rizkallah, M., Saad, R. & Aziz, R. The Human Microbiome Project, Personalized Medicine and the Birth of Pharmacomicrobiomics. Curr Pharmacogenomics Person Med 8, 182–193 (2010).
- Tian, J. & Ma, J. The Value of Microbes in Cancer Neoantigen Immunotherapy. Pharmaceutics 15, 2138 (2023).
- Boesch, M. et al. Tumour neoantigen mimicry by microbial species in cancer immunotherapy. Br J Cancer 125, 313–323 (2021).
- Wilson, I. D. & Nicholson, J. K. Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Translational Research 179, 204–222 (2017).
- Fuller, A. T. IS p-AMINOBENZENESULPHONAMIDE THE ACTIVE AGENT IN PRONTOSIL THERAPY ? The Lancet 229, 194–198 (1937).
- Gingell, R., Bridges, J. W. & Williams, R. T. The Role of the Gut Flora in the Metabolism of Prontosil and Neoprontosil in the Rat. Xenobiotica 1, 143–156 (1971).
- Mehta, R. S. et al. Gut microbial metabolism of 5-ASA diminishes its clinical efficacy in inflammatory bowel disease. Nat Med 29, 700–709 (2023).
- NCBI. https://www.ncbi.nlm.nih.gov/books/NBK556025/#:~:text=Mechanism%20of%20Action,-Digoxin%20has%20two&text=Digoxin%20induces%20an%20increase%20in,effects%20on%20the%20AV%20node.
- Li, H., He, J. & Jia, W. The influence of gut microbiota on drug metabolism and toxicity. Expert Opin Drug Metab Toxicol 12, 31–40 (2016).
- MedlinePlus. https://medlineplus.gov/spanish/druginfo/meds/a601068-es.html.
- van Kessel, S. P. et al. Gut bacterial tyrosine decarboxylases restrict levels of levodopa in the treatment of Parkinson’s disease. Nat Commun 10, 310 (2019).
- Lee, W. Y., Yoon, W. T., Shin, H. Y., Jeon, S. H. & Rhee, P.-L. Helicobacter pylori infection and motor fluctuations in patients with Parkinson’s disease. Movement Disorders 23, 1696–1700 (2008).
- Pierantozzi, M. et al. Helicobacter pylori eradication and l-dopa absorption in patients with PD and motor fluctuations. Neurology 66, 1824–1829 (2006).
- Pierantozzi, M. et al. Helicobacter pylori‐induced reduction of acute levodopa absorption in parkinson’s disease patients. Ann Neurol 50, 686–687 (2001).
- Hashim, H. et al. Eradication of Helicobacter pylori Infection Improves Levodopa Action, Clinical Symptoms and Quality of Life in Patients with Parkinson’s Disease. PLoS One 9, e112330 (2014).
- Goldin, B. R., Peppercorn, M. A. & Goldman, P. Contributions of host and intestinal microflora in the metabolism of L-dopa by the rat. J Pharmacol Exp Ther 186, 160–6 (1973).
- Maini Rekdal, V., Bess, E. N., Bisanz, J. E., Turnbaugh, P. J. & Balskus, E. P. Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science (1979) 364, (2019).
- Spanogiannopoulos, P. et al. Host and gut bacteria share metabolic pathways for anti-cancer drug metabolism. Nat Microbiol 7, 1605–1620 (2022).
- Wallace, B. D. et al. Alleviating Cancer Drug Toxicity by Inhibiting a Bacterial Enzyme. Science (1979) 330, 831–835 (2010).
- Wallace, B. D. et al. Structure and Inhibition of Microbiome β-Glucuronidases Essential to the Alleviation of Cancer Drug Toxicity. Chem Biol 22, 1238–1249 (2015).
- Lin, H.-Y. et al. Entropy-driven binding of gut bacterial β-glucuronidase inhibitors ameliorates irinotecan-induced toxicity. Commun Biol 4, 280 (2021).
- Peters, B. A. et al. The Microbiome in Lung Cancer Tissue and Recurrence-Free Survival. Cancer Epidemiology, Biomarkers & Prevention 28, 731–740 (2019).
- Riquelme, E. et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell 178, 795-806.e12 (2019).
- Hermida, L. C., Gertz, E. M. & Ruppin, E. Predicting cancer prognosis and drug response from the tumor microbiome. Nat Commun 13, 2896 (2022).
- Nejman, D. et al. The human tumor microbiome is composed of tumor type–specific intracellular bacteria. Science (1979) 368, 973–980 (2020).
- Choy, A. T. F. et al. The microbiome of pancreatic cancer: from molecular diagnostics to new therapeutic approaches to overcome chemoresistance caused by metabolic inactivation of gemcitabine. Expert Rev Mol Diagn 18, 1005–1009 (2018).
- Geller, L. T. et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science (1979) 357, 1156–1160 (2017).
- He, Y. et al. Gut microbial metabolites facilitate anticancer therapy efficacy by modulating cytotoxic CD8+ T cell immunity. Cell Metab 33, 988-1000.e7 (2021).
- Encarnação, J. C. et al. Butyrate, a dietary fiber derivative that improves irinotecan effect in colon cancer cells. J Nutr Biochem 56, 183–192 (2018).
- Ghosh, S. et al. Microbial metabolite restricts 5-fluorouracil-resistant colonic tumor progression by sensitizing drug transporters via regulation of FOXO3-FOXM1 axis. Theranostics 12, 5574–5595 (2022).
- Mirzaei, S., Iranshahy, M., Gholamhosseinian, H., Matin, M. M. & Rassouli, F. B. Urolithins increased anticancer effects of chemical drugs, ionizing radiation and hyperthermia on human esophageal carcinoma cells in vitro. Tissue Cell 77, 101846 (2022).
- Clayton, T. A., Baker, D., Lindon, J. C., Everett, J. R. & Nicholson, J. K. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proceedings of the National Academy of Sciences 106, 14728–14733 (2009).
- Klünemann, M. et al. Bioaccumulation of therapeutic drugs by human gut bacteria. Nature 597, 533–538 (2021).
- Đanić, M. et al. Bioaccumulation and biotransformation of simvastatin in probiotic bacteria: A step towards better understanding of drug-bile acids-microbiome interactions. Front Pharmacol 14, (2023).
- Roy, S. & Trinchieri, G. Microbiota: a key orchestrator of cancer therapy. Nat Rev Cancer 17, 271–285 (2017).
- Vitali, F. et al. Early melanoma invasivity correlates with gut fungal and bacterial profiles. British Journal of Dermatology 186, 106–116 (2022).
- Vétizou, M. et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science (1979) 350, 1079–1084 (2015).
- Chaput, N. et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Annals of Oncology 28, 1368–1379 (2017).
- Dubin, K. et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat Commun 7, 10391 (2016).
- Gopalakrishnan, V. et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science (1979) 359, 97–103 (2018).
- Matson, V. et al. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science (1979) 359, 104–108 (2018).
- Routy, B. et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science (1979) 359, 91–97 (2018).
- Gosálbez, L. Microbiome-based diagnostics and biomarkers: Changing the paradigm in microbiology and medicine. https://www.microbiometimes.com/microbiome-based-diagnostics-and-biomarkers-changing-the-paradigm-in-microbiology-and-medicine/. Microbiome Times (2021).
- Pierrard, J. & Seront, E. Impact of the gut microbiome on immune checkpoint inhibitor efficacy-a systematic review. Curr Oncol 26, 395–403 (2019).
- Mohiuddin, J. J. et al. Association of Antibiotic Exposure With Survival and Toxicity in Patients With Melanoma Receiving Immunotherapy. JNCI: Journal of the National Cancer Institute 113, 162–170 (2021).
- Smith, M. et al. Gut microbiome correlates of response and toxicity following anti-CD19 CAR T cell therapy. Nat Med 28, 713–723 (2022).
- Statista. Research and development expenditure of total U.S. pharmaceutical industry from 1995 to 2022. https://www.statista.com/statistics/265085/research-and-development-expenditure-us-pharmaceutical-industry/.
- Matthew Herper. The Cost Of Creating A New Drug Now $5 Billion, Pushing Big Pharma To Change. https://www.forbes.com/sites/matthewherper/2013/08/11/how-the-staggering-cost-of-inventing-new-drugs-is-shaping-the-future-of-medicine/?sh=78ad7a4c13c3.
- Zhavoronkov, A. $6.16 Billion Per Drug Approval: Almost Half of Big Pharma Companies Hit Negative R&D Productivity. Forbes (2023).
- Lowe, D. Eroom’s Law. https://www.science.org/content/blog-post/eroom-s-law.
- Biotechnology Innovation Organization. Clinical Development Success Rates and Contributing Factors 2011–2020. https://go.bio.org/rs/490-EHZ-999/images/ClinicalDevelopmentSuccessRates2011_2020.pdf. (2021).
- Kola, I. & Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 3, 711–716 (2004).